Usage
TrioMix requires the input of sequence alignment file (BAM or CRAM files) of trios and a reference FASTA file. SNP BED file can be used as an optional argument.
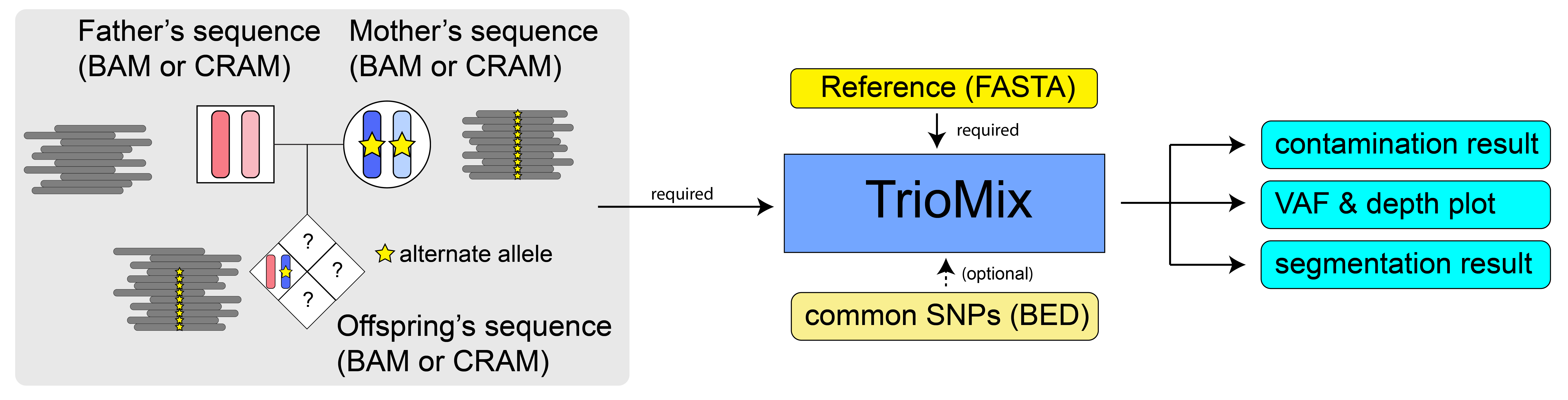
Basic TrioMix command line: Detection of intrafamilial contamination in the offspring
By default, TrioMix uses the parental genotypes (GroupA, B, C SNPs) to infer the intrafamilial contamination level in the offspring. Since -o is commonly reserved for outputs, we use -c, --child to refer to the offspring. The basic command line of using TrioMix is the following:
$ python triomix.py -f father.bam -m mother.bam -c child.bam -r reference.fasta
TrioMix command line with common SNP only
Using a pre-selected list of common SNP would speed up the total runtime of TrioMix as the computation is limited to those regions instead of the entire genome. TrioMix provides a list of common GRCh38 and GRCh37 SNPs selected from the GnomAD database. These two files are included in the github repository as a common_snp folder. A -s argument specifies the SNP database that can be used. User can provide one’s own set of SNP in BED format.
$ python triomix.py -f father.bam -m mother.bam -c child.bam -r reference.fasta -s common_snps/grch38_common_snps.bed.gz
Command line arguments
$ python triomix.py -h
usage: triomix [-h] [--version] -f FATHER -m MOTHER -c CHILD -r REFERENCE [-s SNP] [-t THREAD] [-o OUTPUT_DIR]
[-p PREFIX] [--runmode {single,joint,all}] [-u {0,1}] [--parent] [-d DOWNSAMPLE]
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
-f FATHER, --father FATHER
Father's BAM or CRAM file
-m MOTHER, --mother MOTHER
Mother's BAM or CRAM file
-c CHILD, --child CHILD
Child's BAM or CRAM file
-r REFERENCE, --reference REFERENCE
Reference FASTA file
-s SNP, --snp SNP Optional list of SNP sites as a BED (or BED.gz) file
-t THREAD, --thread THREAD
Multithread to utilize. Default=1
-o OUTPUT_DIR, --output_dir OUTPUT_DIR
Output directory. Default=current working directory
-p PREFIX, --prefix PREFIX
prefix for the output file. If not specified, will use the SM tag from the child bam's
header
--runmode {single,joint,all}
Runmode for mle.R script. 'single' assumes only 1 contamination source within family.
'joint' calculates the fraction of all family members jointly. 'all' runs both modes.
Default=all
-u {0,1}, --upd {0,1}
0: mle will filter out vaf=0 or 1 in sites where parental genotypes are homo-ref + homo-alt
(GroupA SNPs) 1: mle will identify UPDs which appears as contamination. Default=1
--parent Run detection of parental DNA contamination with child's DNA
-d DOWNSAMPLE, --downsample DOWNSAMPLE
Downsampling for plotting.
Default output files
TrioMix produces several output files files.
*.x2a.depth.tsv: contains the depth ratio chrX vs autosome of each individual in a trio. Males are expected to have ~0.5 while female should have value ~1.0.
*.child.counts: contains the position of the SNP loci in either GroupA, B, or C. Contains the read depths, alternative read counts for the trios. In addition, based on the parental genotype, will determine whether the child inherited the SNP from the father (F) or the mother (M). This file is used as the input for mle.R which estimates the contamination level using maximum likelihood estimation.
*.child.counts.upd.segments.tsv: contains the VAF values for GroupA SNPs that have been segmented for UPD analysis
*.child.counts.plot.pdf: visualization of depth and VAF plots of GroupA and GroupB SNPs in the child.
*.child.counts.summary.tsv: contains the final estimated values of contamination from various sources in the child. Detailed information on each column is as follows.
child_contam_by_sibling_joint # contamination estimated from joint analysis of all family members (GroupA + GroupB used)
child_contam_by_father_joint # contamination estimated from joint analysis of all family members (GroupA + GroupB used)
child_contam_by_mother_joint # contamination estimated from joint analysis of all family members (GroupA + GroupB used)
convergence_joint # mle function convergence status. If 0, then indicates convergence succeeded.
child_contam_by_sibling # contamination estimated assuming only sibling contaminating (GroupB used)
child_contam_by_father # contamination estimated assuming only father contaminating (GroupA used)
child_contam_by_mother # contamination estimated assuming only mother contaminating (GroupA used)
groupA_father # number of paternal GroupA variants identified
groupA_mother # number of maternal GroupA variants identified
groupB_father # number of paternal GroupB variants identified
groupB_mother # number of maternal GroupB variants identified
denovo_error_rate # fraction of alternative read count at GroupC SNPs
TrioMix with whole-exome sequencing
TrioMix can be used with whole-exome sequencing. In this case, we recommend running the command without the -s common_snp/common_snps.bed.gz to capture rare SNPs as well. This increases the overall number of SNPs while having minimal effect on the computational time due to smaller target in the exome sequeincing. For plotting, using -d 1 is recommended to capture all data points in the plot without downsampling.
$ python triomix.py -f father.bam -m mother.bam -c child.bam -r reference.fasta -d 1
Detection of intrafamilial contamination in the parent (i.e. parent DNA contaminated by child, or by another parent)
To detect intrafamilial DNA contamination in the parent, --parent option can be used. This will use GroupD SNPs (where offspring’s genotype is homo-alt) to detect the offspring DNA contaminating in the parents.
$ python triomix.py -f father.bam -m mother.bam -c child.bam -r reference.fasta -s common_snps/grch38_common_snps.bed.gz --parent
Additional output generated with --parent
*.parent.counts: contains the position of the SNP loci in either Group D or E. Contains the read depths, alternative read counts for the trios. This file is used as the input for mle_parent.R which estimates the contamination level using maximum likelihood estimation.
*.parent.counts.plot.pdf: visualization of depth and VAF plots of GroupD and GroupE SNPs in the parents.
*.parent.counts.summary.tsv: contains the final estimated values of contamination from various sources in each parents. Detailed information on each column is as follows.
mother_contam_by_child # contamination estimated in the mother (GroupD)
father_contam_by_child # contamination estimated in the father (GroupD)
mother_contam_by_father # contamination estimated in the mother (GroupE)
father_contam_by_mother # contamination estimated in the fother (GroupE)
groupD_mother # number of maternal GroupD variants identified
groupD_father # number of paternal GroupD variants identified
groupE_mother # number of maternal GroupE variants identified
groupE_father # number of paternal GroupE variants identified
Running TrioMix with a docker image
Following example demonstrates how docker image can be used for runnint TrioMix.
# Download docker image from dockerhub
$ VERSION=v0.0.1 # download specific release version tag of TrioMix
$ docker pull cjyoon/triomix:$VERSION
# Run triomix with docker image
$ docker run \
-v /path/to/bamfile:/path/to/bamfile \ # bind all folders where input files are located
-v /path/to/reference:/path/to/reference/ \
-v /path/to/output_dir:/path/to/output_dir \ # also bind the location of output folder
-it cjyoon/triomix:$VERSION \
python /tools/triomix/triomix.py \ # location of triomix.py in the docker image
-f /path/to/bamfile/father.bam \ # location of father's bam file
-m /path/to/bamfile/mother.bam \ # location of mother's bam file
-c /path/to/bamfile/mother.bam \ # location of child's bam file
-s /tools/triomix/common_snp/grch38_common_snp.bed.gz \ # location of common SNP file in the docker image
-r /path/to/reference/reference.fa \ # location of reference FASTA file
-o /path/to/output_dir # location where output files are saved