Example
Example workflow
Contamination can be simulated by randomly selecting read sequences from two BAM (or CRAM) files at a defined ratio. The total number of reads in each bam file is adjusted when creating a subsampled BAM file which is later merged into a single ‘contaminated’ bam file.
Test run with 1000 genomes trio
In our github a test script test.sh is provided which can created a simulated contamination BAM files from a 1000 genomes family.
$ sh test.sh
This will download the 1000 genomes trio CRAM files, create simulated contamination files by subsampling. Note this will requires samtools to be installed in your $PATH. Below, a few example cases in the test.sh are described.
Download a 1000 genomes trio (+ a sibling) CRAM files
The following command downloads a 1000 genomes trio (+ a sibling) into current working directory
# download M008 family's WGS from 1000 genomes project ftp.
# proband
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3989418/NA19662.final.cram -O proband.cram
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3989418/NA19662.final.cram.crai -O proband.cram.crai
# father
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR323/ERR3239902/NA19661.final.cram -O father.cram
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR323/ERR3239902/NA19661.final.cram.crai -O father.cram.crai
# mother
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3989417/NA19660.final.cram -O mother.cram
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3989417/NA19660.final.cram.crai -O mother.cram.crai
# sibling
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3989425/NA19685.final.cram -O sibling.cram
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3989425/NA19685.final.cram.crai -O sibling.cram.crai
# download the reference fasta file
wget -nc https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.fasta
samtools faidx Homo_sapiens_assembly38.fasta
Offspring DNA contaminated by mother’s DNA
The following scripts create a simulated contamination consisting of 75% offspring’s DNA + 25% mother’s DNA. SCRIPTPATH should be set to the folder where triomix.py is located.
SCRIPTPATH=/path/to/triomix # change this to triomix's github folder path. This is automatically detected in test.sh
#################################
# offspring contaminated by mother simulation
python $SCRIPTPATH/simulate_familial_mixture.py \
-f father.cram \
-m mother.cram \
-c proband.cram \
-s sibling.cram \
-r 0 0.25 0.75 0 -o offspring75_mother25 # 0% father, 25% mother, 75% offspring 0% sibling
# run TrioMix on offspring contaminated by mother
python $SCRIPTPATH/triomix.py \
-f father.cram \
-m mother.cram \
-c offspring75_mother25/familymix.bam \
-r Homo_sapiens_assembly38.fasta -t 8 \
-s $SCRIPTPATH/common_snp/grch38_common_snp.bed.gz \
-p offspring75_mother25 -o results
This will produce the contamination estimation file offspring75_mother25.child.counts.summary.tsv
$ cat offspring75_mother25.child.counts.summary.tsv
type value
child_contam_by_sibling_joint 1.8873588344360515e-4
child_contam_by_father_joint 8.167772506667559e-5
child_contam_by_mother_joint 0.2507938532424764
convergence_joint 0
child_contam_by_sibling 0.2533023916508015
child_contam_by_father 6.474095861474213e-9
child_contam_by_mother 0.2507121759058531
groupA_father 73041
groupA_mother 73077
groupB_father 125753
groupB_mother 124975
denovo_error_rate 3.992565674974727e-4
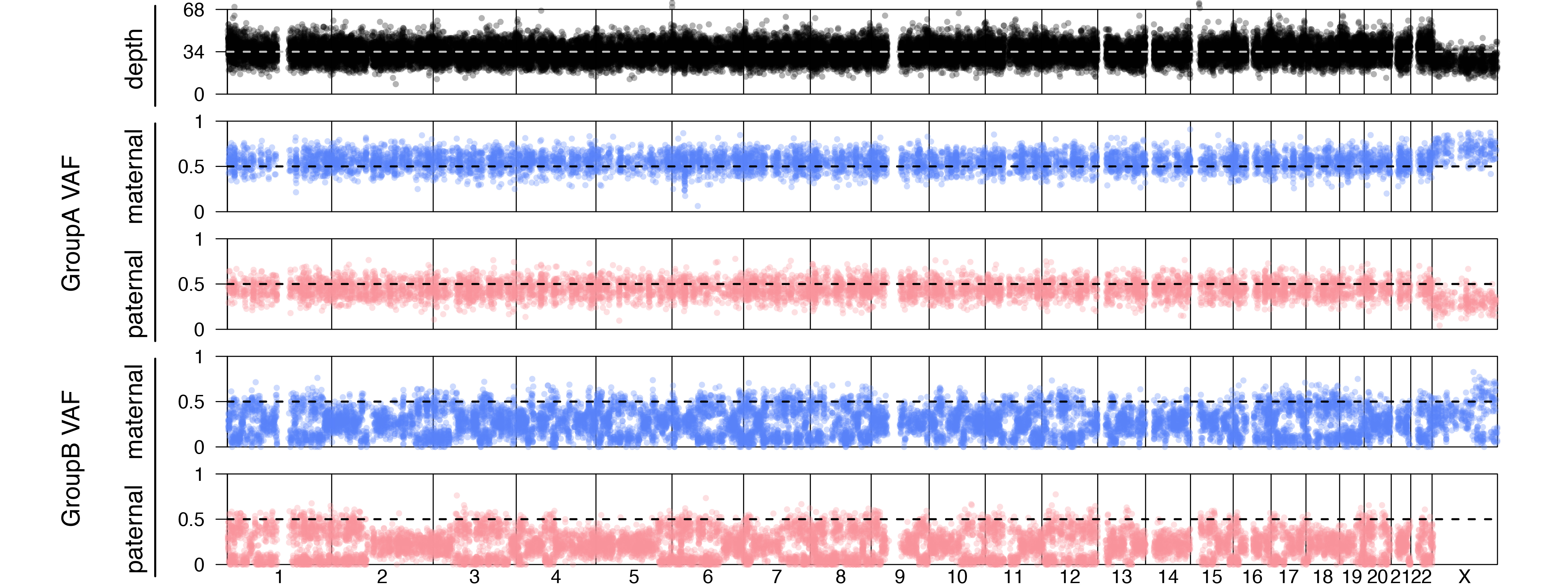
Joint estimation assuming all possible contamination from all family members estimated 25% contamination only from the mother. However, if we look at the estimation assuming only one individual at a time, fitting the same data may show maximum likelihood with 25% contamination by the father and also 25% contamination of the mother. Thus, the joint method provides the most definitive contamination estimation.
A genome wide plot for this simulated case is shown.
Offspring DNA contaminated by father, mother, and a sibling
Here, we simulate a complex case where the offspring’s DNA is coontaminated by the father, mother, and a sibling simultaneously.
##################################
# multiple contamination simulation, father=10%, mother=20%, offspring=40%, sibling=30%
python $SCRIPTPATH/simulate_familial_mixture.py \
-f father.cram \
-m mother.cram \
-c proband.cram \
-s sibling.cram \
-r 0.10 0.20 0.40 0.30 -o complexmix # 10% father, 20% mother, 40% offspring 30% sibling
# run TrioMix on the complex contaminated case
python $SCRIPTPATH/triomix.py \
-f father.cram \
-m mother.cram \
-c complexmix/familymix.bam \
-r Homo_sapiens_assembly38.fasta -t 8 \
-s $SCRIPTPATH/common_snp/grch38_common_snp.bed.gz \
-p complexmix -o results
This will produce the contamination estimation file complexmix.child.counts.summary.tsv
$ cat complexmix.child.counts.summary.tsv
type value
child_contam_by_sibling_joint 0.2900164906231494
child_contam_by_father_joint 0.09759458292835317
child_contam_by_mother_joint 0.2006493758684508
convergence_joint 0
child_contam_by_sibling 0.29732892004146544
child_contam_by_father 8.909208856139373e-9
child_contam_by_mother 0.10305479295638349
groupA_father 73041
groupA_mother 73077
groupB_father 125753
groupB_mother 124975
denovo_error_rate 3.1783417104182737e-4
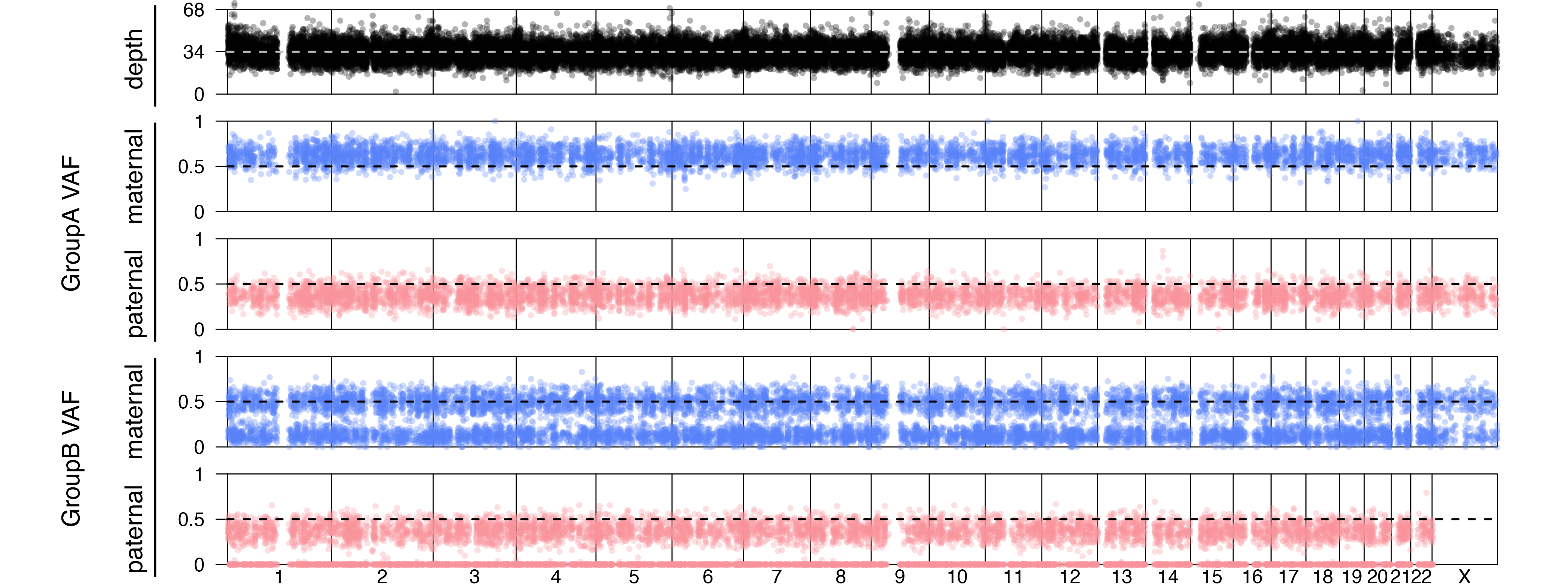
Joint estimation of all family members accurately estimated the 10% father’s contamination, 20% mother’s contamination, and 30% sibling’s contamination in the offspring’s DNA. In the single contamination estimation mode, only the difference between the father and mother is measured at 10%.
A genome wide plot for this simulated case is shown.